
Amyloid isn’t Alzheimer’s – How NOT to Study a Disease
by Patrick Holford
Alzheimer’s disease has, for decades, been framed primarily as a problem of amyloid plaques building up in the brain.
It is a compelling idea. It is measurable, visible on brain scans, and has shaped billions in research funding and drug development. There’s just one problem.
It doesn’t adequately explain what we see in people.
Many individuals with significant amyloid in their brains remain cognitively normal. Others develop clear dementia with little or no amyloid present (1). Treatments that successfully reduce amyloid have, at best, produced only modest effects on slowing decline, with no meaningful reversal of symptoms (2).
Amyloid is associated with Alzheimer’s disease, but association is not the same as causation.
And that distinction changes everything.
This growing tension between theory and evidence is explored in depth by neurobiologist Karl Herrup in How Not to Study a Disease, where he challenges how the field has come to define and pursue Alzheimer’s.
Some people develop increasingly severe cognitive decline as they age. This affects roughly one in ten older adults. We call this dementia. In some cases, brain scans show clear shrinkage in key regions, particularly the medial temporal lobe, which is then used to diagnose Alzheimer’s disease. So we have two observable features: a decline in cognitive function and measurable loss of brain tissue.
The question that follows is simple, but critical.
What is actually causing this process?
What causes Alzheimer’s?
Amyloid has long been positioned as the answer. Yet the evidence tells a more complicated story.
A significant proportion of older adults have amyloid plaques in their brains and remain cognitively normal. At the same time, some individuals with clear dementia show little or no amyloid pathology. Amyloid is associated with Alzheimer’s, but that is not the same as being the cause.
If amyloid were the primary driver, then reducing it should meaningfully change the course of the disease. However, interventions designed to reduce amyloid have consistently lowered amyloid burden in the brain, yet produced, at best, modest effects on slowing decline, often measured as very small changes on cognitive scales. There has been no meaningful reversal of symptoms. In some cases, safety concerns have emerged, including brain bleeding and swelling.
Recent large-scale evidence (6) now reinforces this point. A 2026 Cochrane review concluded that although anti-amyloid monoclonal antibodies can remove amyloid from the brain, this does not appear to translate into clinically meaningful effects for people with mild cognitive impairment or mild dementia due to Alzheimer’s disease, while increasing the risk of amyloid-related imaging abnormalities.
From a scientific perspective, this adds to an already substantial body of evidence suggesting that amyloid accumulation, on its own, does not explain the disease process. It may be part of the picture, but it is not the engine driving it.
And yet, the field has remained heavily focused on this single pathway.
When a hypothesis becomes a lens
Research tends to follow what is measurable, fundable, and already established. Over time, this can narrow the lens rather than expand it.
Amyloid has become that lens. As Karl Herrup argues in his book, once a hypothesis becomes dominant, it can begin to shape not just what is studied, but how results are interpreted and what gets funded next.
The result is that vast resources have been invested in understanding and modifying amyloid biology, while other avenues have received comparatively less attention. We have learned a great deal about amyloid itself, but we are not significantly closer to preventing or reversing the condition that matters most to patients, which is cognitive decline.
This is not unusual in science. Once a model becomes dominant, it shapes the direction of funding, research questions, and even how people interpret results.
A similar pattern is now emerging with another biomarker, p-tau.
Tau is a normal protein that, under certain conditions, becomes altered and associated with the tangles seen in Alzheimer’s pathology. Higher levels of p-tau are linked with increased risk, but again, association does not establish causation. Many individuals have elevated levels without clinical symptoms.
As with amyloid, the risk is that a marker becomes mistaken for the mechanism.
What does influence the disease process?
This is where the picture becomes more interesting.
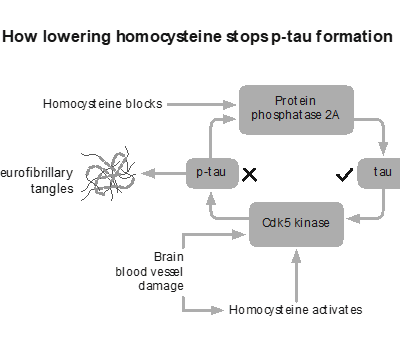
There are factors that sit upstream of both amyloid and tau, influencing the environment in which brain cells function or fail. One of the most studied is homocysteine, a marker of methylation and B vitamin status.
Unlike amyloid, intervention trials have shown that homocysteine influences outcomes. In the VITACOG study, lowering elevated homocysteine with targeted B vitamins significantly slowed the rate of brain atrophy in individuals with mild cognitive impairment, with corresponding effects on cognitive decline. This is much closer to what we would describe as a disease-modifying effect (3,4). It also raises questions explored in Apparently healthy but diagnosed with Alzheimer’s about whether current diagnostic models are identifying true disease drivers or simply biomarkers.
That does not mean homocysteine is the only cause. It is one piece of a larger system. But it illustrates an important point. When you influence the underlying biology of the brain, rather than a downstream marker, you begin to see meaningful change.
For more on this approach, see our article on early diagnosis of Alzheimer’s: amyloid protein vs homocysteine testing.
A systems problem, not a single cause
Alzheimer’s does not behave like a single-cause disease.
It is better understood as the result of multiple interacting processes. These include inflammation, oxidative stress, insulin resistance, mitochondrial dysfunction, and impaired methylation, among many others (5). Each of these affects how brain cells are built, maintained, and powered.
Individually, they may not be sufficient to cause disease. Together, they can create the conditions in which the brain becomes vulnerable.
This is closer to how we understand most chronic conditions. Not as a single fault, but as a convergence of pressures that eventually exceed the system’s ability to compensate. A more useful way to think about it is not as one switch flipping, but as several dials turning in the wrong direction at the same time.
Why this matters
If we continue to focus primarily on downstream markers such as amyloid or p-tau, we risk missing the broader picture.
If instead we look at the upstream drivers, the factors that influence brain structure, function, and energy supply, we open up a different set of possibilities. Not just for treatment, but for prevention.
These are not fringe ideas. They are part of a growing shift in how Alzheimer’s is being understood, questioned, and re-examined. Researchers like Karl Herrup are helping to bring that conversation into the open, challenging long-held assumptions and asking more useful questions about what truly drives the disease.
It’s a conversation that is only just beginning to reach wider audiences.
This is exactly what we’ll be exploring at Alzheimer’s Prevention: New Frontiers conference, where leading researchers and clinicians will come together to look beyond single-cause models and towards a more complete understanding of brain health and cognitive decline.
Is prevention the real solution?
The prevailing model in medicine has been to identify a single cause and target it with a treatment. That works well for some conditions. It is less suited to complex, multifactorial diseases like Alzheimer’s.
A systems-based approach asks a different question.
What combination of factors leads to decline, and how do we shift that combination in the opposite direction?
This is the approach we take at Food for the Brain. By combining cognitive testing with blood testing and lifestyle information, it becomes possible to see patterns, not just isolated variables. Over time, this allows us to understand what drives resilience as well as risk.
It is likely that we will not find a single primary cause of Alzheimer’s. What we may find is something more useful: a set of modifiable factors that, together, determine whether the brain maintains function or begins to decline.
In that sense, prevention may not just be part of the solution.
It’s the solution.
Find out more with Alzheimer’s: Prevention is the Cure book here
Learn more about our Alzheimer’s Prevention: New Frontiers Conference here.
References
- Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562.
- van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9–21.
- Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment. PLoS One. 2010;5(9):e12244.
- de Jager CA, Oulhaj A, Jacoby R, et al. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment. Int J Geriatr Psychiatry. 2012;27(6):592–600.
- Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413–446.
.
Thank you for reading!
Food for the Brain is a non-for-profit educational and research charity that offers a free Cognitive Function Test and assesses your Dementia Risk Index to be able to advise you on how to dementia-proof your diet and lifestyle.
By completing the Cognitive Function Test you are joining our grassroots research initiative to find out what really works for preventing cognitive decline. We share our ongoing research results with you to help you make brain-friendly choices.
Please support our research by becoming a Friend of Food for the Brain.