By Patrick Holford
This is taken from Patrick Holford’s NEW book – Alzheimer’s: Prevention is the Cure which you can pre-order here today!
If you think that Alzheimer’s or dementia is caused by amyloid plaques in the brain, or tangles of nerves (neurofibrillary tangles) associated with p-tau, you have been successfully fooled. But you would not be alone. There is a vast sleight of hand going on that remarkably continues to hijack research into true causes and potential cures for Alzheimer’s, despite a mountain of clear evidence to the contrary.
Let’s start at the beginning. Some people suffer increasingly severe cognitive decline. This affects about one in ten older people. We call this dementia. Some people with dementia, on scanning their brains, have big gaps in the central part of the brain. This is used to diagnose the form of dementia known as Alzheimer’s disease, due to the clear evidence of ‘pathology’ – something wrong in the brain that amounts to the death of significant amounts of brain cells in critical areas.
So, here we have two clear diagnostic criteria. Firstly, a loss of cognitive function, which is what is tested by the dementia prevention charity, Food for the Brain, with its free, online Cognitive Function Test. Secondly, a loss of actual brain, which is diagnosed by a type of brain scan of the central or medial part of the brain. This scan was first developed based on research at Oxford University, headed by Professor David Smith, who is a member of the Scientific Advisory Board – see foodforthebrain.org/sab.
So, then the question is: what causes it? There has never been any evidence (and there is still no evidence), that Alzheimer’s, except for the very rare early-onset types of Alzheimer’s caused by genes, is caused by deposits of amyloid protein or amyloid plaque in the brain. “Over the past 25 years, Alzheimer’s research has suffered a litany of ostensible fraud and other misconduct by world-famous researchers and obscure scientists alike, all trying to ascend in a brutally competitive field,” claims Charles Piller in the New York Times [1], author of the book, Doctored: Fraud, Arrogance, and Tragedy in the Quest to Cure Alzheimer’s.
An example of the ‘doctoring’, reported by Dr Matthew Schrag, professor of neuroscience at Vanderbilt University, in Science in 2022, identified as many as 10 papers on the protein that deserve deeper scrutiny [2]. The report also cited other prominent researchers who have had difficulty replicating results of the studies on the specific proteins. The original research has now been withdrawn.
The reality is that about 30% of older people have plaques in their brains without dementia. About 15% of those with dementia don’t have amyloid plaques [3]. Having amyloid plaques doesn’t cause dementia. Mice whose brains have been molecularly engineered to produce amyloid plaques behave normally. Even a head full of plaques only results in mild memory problems. Many of us have plaques in our brain and remain completely healthy.
Less than 1% of diagnoses of Alzheimer’s are caused by genes. These account for very early onset cases. The genes are Amyloid Protein Precursor (APP) gene and Presenilin (PS1 and PS2). Now these rare dementias can all plausibly be assumed to be caused by amyloid plaque deposition, and to be potentially curable by its removal. However, being so rare, there is little commercial imperative to find out. There is, however, one study in 2019 that tested two different anti- amyloid treatments given to those with this rare early-onset Alzheimer’s. Despite both drugs effectively lowering the amyloid burden, there was no clinical improvement, but a slight worsening for one of these treatments compared to placebo. In addition, one in five had brain swelling [4]. That would be reason enough to give up on the amyloid hypothesis.
The big mistake, however, was the leap of faith assuming that therefore ALL Alzheimer’s, which makes up two thirds of dementias, were also caused by amyloid accumulation and could be so treated with drugs to lower the amyloid burden.
Blocking the enzymes that make amyloid has made people worse, not better, despite lessening the amyloid burden [5]. Vaccinating animals to remove the plaques doesn’t change anything to do with dementia, but it does reduce the amyloid. The anti-amyloid vaccine injections in humans have been equally ineffective (in terms of impacting dementia), despite lowering their amyloid burden.
The pharmaceutical companies running these failed trials have pushed and pushed until they could just about get a ‘significant’ difference in the rate of degeneration of patients versus placebo on assessment questionnaires – just enough to get a medical licence despite being clinically ineffective. Lecanemab was the first to be licensed in the UK, in 2024. The difference in the lecanemab trial between those on the drug and those on placebo was equivalent to less than half a point (0.45) change on an 18 point Clinical Dementia Rating (CDR) scale [6]. According to a British Medical Journal editorial this decrease “fell well short, representing only around a third of what a minimum clinically important difference might look like.” [7]
On another scale, the Alzheimer’s Disease Assessment Scale (ADAS), both those on placebo and drug treatment start to decline rapidly after 15 month (see figure 1 right). The Alzheimer’s Society [8] report this miniscule difference as ‘Lecanemab slowed down the speed at which memory and thinking skills got worse by 27%’.
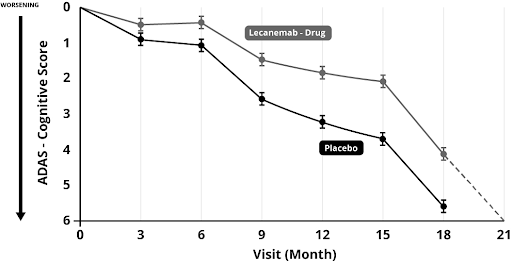
This was the figure reported in the newspapers, ignoring the fact that, in reality, those on the drug just hit the same rock bottom about 3 months later than those on the placebo and the difference is so small that no-one is likely to notice. No-one got better. They all got worse. Quite a few got adverse effects, with brain bleeding and swelling. More than a quarter had adverse reactions. A few died as a consequence. Is three months of ‘slightly less worse’ worth the suffering of one in four and the death of a few (about one in 500) at vast expense? If such treatment was started before a person was put into care, at best it could mean putting them in a care home three months later, potentially saving £3,000. If treatment were given while in a care home it would mean three months more time in a care home, potentially costing £3,000 more. Either way, at a treatment cost more likely to be in the region of £50,000 per year this is clearly not cost effective for the NHS.
But still, drug regulatory agencies, paid for by the drug industry, dished out licences because the results were ‘statistically significant’- the result of enrolling as many as 1795 people. Larger trials make small positive results look better.
Even so, the UK watchdog NICE said the evidence wasn’t good enough and recommended the National Health Service not to give anti-amyloid treatment – at about £50,000 a patient per year when you factor in the cost of scans needed to check for bleeding and swelling with each injection and medical costs. Despite this you’ll read newspaper headlines such as ‘Alzheimer’s drugs should be prescribed like statins’, as appeared in the Telegraph [9], interviewing Professor Hardy from UCL.
Please also bear in mind that even these bad results are the best that the drug company, who funded and ran their own drug trial could conjure up with questionable methodology. The CDR (Clinical Dementia Rating) is essentially a questionnaire completed by a partner/carer and clinician. If you had vested hope that your loved one might improve on an experimental drug, might you answer slightly more positively?
Also, these trials are meant to be ‘double-blind’ i.e., the patient (and carer/partner) doesn’t know if they are injected with a placebo or drug. But when almost a quarter get severe side-effects just how ‘double-blind’ is it? If you got side-effects, assumed you were on the drug, would the hope of improvement bias your answers?
The truth is it is easy to cheat in trials, or at least massage the results in your favour, and there is a strong motive to do so, if it’s your drug, job and profits at stake. That’s why I trust trials done on drugs or vitamins by independent researchers. These don’t exist for the anti- amyloid drugs and are unlikely to, due to the vast expense of such trials. Independent researcher Sarah Ackley wanted to do a meta-analysis for publication in the British Medical Journal of anti-amyloid drug trials. She identified 34 trials suitable for inclusion in her analysis, but was denied access to the data of 20.[10] In other words, a drug company can run a failed trial, ditch it and move on, only revealing those that show an effect. Out of the 14 trials she was allowed to see the data from their meta-analysis concluded: “Combined results from 14 randomized controlled trials provide evidence that reduction in amyloid levels alone is unlikely to substantially slow cognitive decline within the follow-up period of most typical trials. The results of pooled estimates suggest that use of anti-amyloid drugs is not a viable strategy for the prevention or treatment of Alzheimer’s disease and that other potential targets may merit more attention.”
Pharmacology Professor David Smith from the University of Oxford responded to the British Medical Journal[11] saying “Scientists should seriously question the validity of the basic amyloid hypothesis, as was pointed out more than 10 years ago in relation to earlier trials.[12] These findings should direct our attention to the prevention of Alzheimer’s disease by slowing down the disease process, for which there are many possible approaches. The study also raises an ethical question: is it justifiable to ask patients to undergo yet more trials of anti-amyloid treatments? Moreover, we should all question the morality of the drug companies that declined to give these researchers access to data for 20 of the 34 trials they wanted to study.”
In scientific terms, the poor results of the clinical trials, despite lowering amyloid burden, added to the already existing mountain of evidence, that amyloid deposits don’t cause Alzheimer’s; lowering it doesn’t stop the disease process, doesn’t improve cognitive powers in any meaningful way and doesn’t slow down brain shrinkage. In fact, if anything, it accelerates the main physical measure of brain shrinkage. In the anti-amyloid trial for donanemab, those on the amyloid treatment had considerably more whole brain shrinkage – greater than 20% more than those on the placebo (see graph below).[13]
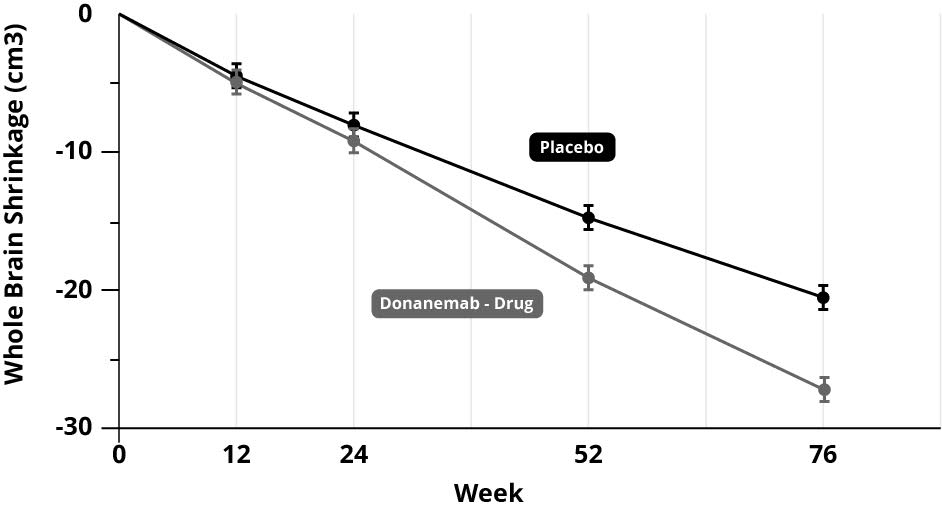
You would think that the whole field would get the message by now; stop funding this dead end and explore other avenues. But there is a lot of investment in the ‘amyloid cascade hypothesis’ that no-one wants to give up. It’s become an unhealthy obsession.
In the US, the Alzheimer’s Association and in the UK the Alzheimer’s Society and Alzheimer’s Research UK, have all supported this line of research and continue to do so. The Alzheimer’s Society, having part funded original research into amyloid with Professor Hardy, consider this their greatest contribution to the field, ‘revolutionising dementia research’.[14] The trouble is, it’s a dead end.
In 2024, there were 164 clinical trials registered assessing 127 drugs, many of which are based on amyloid and p-tau.[15] With several million ‘eligible’ patients, pharma is not going to give up.
It’s partly to do with money. No-one can get research money if they’re not looking at amyloid (or p-tau – more on this in a minute).
In the UK, the Medical Research Council continues to pour good money after bad by making another £20 million available for drug trials.[16] That’s taxpayers’ money backing the wrong horse, despite a lousy track record. In the US, the National Institutes of Health and the National Institutes of Aging spend vast sums pushing in this fruitless direction. Big pharma spends twice as much as the government agencies and the charities, both of which are funded by the taxpayers, probably around $150 billion so far. So, perhaps $250 billion has been spent getting almost nowhere. Sure, we know a lot more about amyloid and p-tau, but are no closer to a ‘cure’.
I remember when, at the G8 Summit in 2013 in London, pharma- funded scientists said, Within ten years we’ll have a cure. Listening to the BBC Radio 4’s Inside Health programme on ‘What’s next for Alzheimer’s’ [17] in November last year, they said the same thing. How can you claim you’ll have a cure when you don’t even know the cause? I predict we’ll be in the same place in ten years if the Alzheimer’s industry doesn’t move on from amyloid and p-tau.
But it gets worse. Despite nothing but evidence to the contrary, based on the completely false notion that ‘Alzheimer’s IS amyloid’, we are being told an amyloid blood test is around the corner. This will tell us nothing useful. It won’t tell us who has Alzheimer’s or who is at risk. So why take the test?
The case for developing a test was made by Professor Hardy in the Telegraph. “My dream is you go the doctor at age 60, have a test, just as you would do for cholesterol. So, it finds you’re at high risk for Alzheimer’s disease, let’s put you on anti-amyloid drugs. Scientifically not difficult.” This may be his pipe dream but, as you have read, no treatment has yet shown a clinically significant effect. A Cognitive Function Test is a better (and free) predictor than an amyloid test, anti- amyloid drugs don’t work and are dangerous. Also, statins don’t work nearly as well as we were led to believe. But the two-step dance of a test that feeds a prescription to healthy people certainly made a lot of money. Over $1 trillion. Statements like this are about sales not science. At best, not that there is evidence to support this, he says that it could
mean going from diagnosis to nursing home in seven years instead of five years. In other words, no-one gets better or stays the same. They would just get worse more slowly.
All this is laid out beautifully in a book by Karl Herrup, Professor of Neurobiology and Investigator at the Alzheimer’s Disease Research Centre at the University of Pittsburgh, called How NOT to study a disease – The Story of Alzheimer’s. If you are questioning what I am saying, please read this book. You can find it in the online bookstore at foodforthebrain.org in the books section.
The medical-pharmaceutical industry is so desperate to find a treatment and make money that it just can’t give up. It reminds me of the story of Mullah Nasrudin, who was looking at the illuminated ground under a lamp post. A passer-by asked, ‘What are you looking for?’ The Mullah said, ‘I dropped a coin.’ The passer-by replied, ‘Did you drop it around here?’ The Mullah said, ‘No, but it’s the only place I can see’.
It’s akin to a campaign to ‘cure’ lemmings when the only cure is for them not to jump off the cliff in the first place. Why spend all that money researching how to give lemmings the medical attention and hospital care as they approach death, when there is a far simpler and less expensive way to help them not need it. Prevention.


A person with dementia will cost the state and family around £100,000 [18]. We can help someone substantially reduce their risk by joining Food for the Brain’s COGNITION programme with a small annual donation. So, for everyone we save from dementia, we could help thousands more.
But let’s be clear. It is true that having lots of amyloid in your brain can increase the PROBABILITY of getting Alzheimer’s in the future, in much the way that being older also increases the probability of getting Alzheimer’s. But it doesn’t cause it. So ‘curing’ amyloid won’t cure the disease.
The same thing is happening with another ‘marker’ in the brain called p-tau, which is associated with having more tangled nerves. Tau is a normal protein that becomes an abnormal, toxic protein called p-tau. The ‘p’ stands for phosphorus or ‘phosphorylated’ because there’s an enzyme that adds on the ‘p’ and another that takes it away. Much like amyloid, having more p-tau increases the PROBABILITY of Alzheimer’s, but does it cause it? Many people have raised levels of p-tau (we all have some) with no problems at all. However, unlike amyloid, there is a threshold such that if you have a lot of p-tau, which means a lot of tangled nerves, this does correlate with the degree of cognitive impairment.
By using the same sleight of hand, £10 million has been put up by the Bill Gates Foundation and people funding Alzheimer’s Research UK, to find the blood test for p-tau (I think they’ve already decided on one called p-tau 217), despite questionable evidence that p-tau causes Alzheimer’s and may just be an artefact of the disease process. In a similar way, tooth decay caused by nutrition and lifestyle deficiencies, such as too much sugar and not brushing your teeth.
No doubt, those with raised p-tau 217 will be told they have ‘pre-clinical dementia’, despite no evidence that they do. Of course, if they had a p-tau lowering drug that actually worked, as in reducing dementia risk, that might be excusable, but they don’t. Those that have blocked the enzymes that cause the accumulation of p-tau have failed. A more relevant question is what causes p-tau to go up? In other words, go to the root of the problem. However, this sleight of hand may be used to sell drugs that don’t work, much like ‘cholesterol’ has been used to sell statins.
My cholesterol is slightly high – I have no disease, no risk factors for heart disease and there is no evidence that lowering my cholesterol will lower my future risk, but still my doctor wants to prescribe me statins. Why? Because GPs are given a calculator called QRisk where you pop in age and cholesterol level and it says ‘prescribe statins’. In any case – two thirds of heart attacks are predicted by high homocysteine – not cholesterol.
With the failure of amyloid drugs to commercialise much emphasis is being put on p-tau inhibitor drugs. So far there are no good clinical results with several horses in the race.
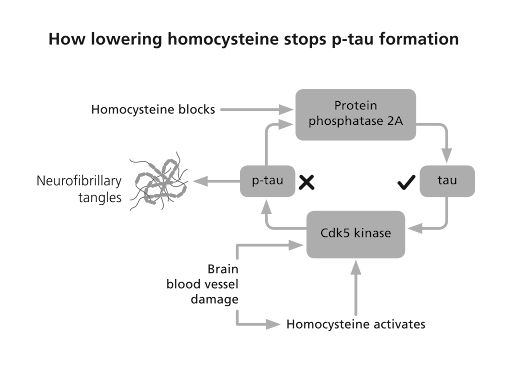
The only thing I know that does lower p-tau is lowering homocysteine with B vitamins. Homocysteine, a toxic amino acid also found in those with Alzheimer’s and dementia, promotes the enzyme that makes p-tau and blocks the enzyme that clears it from your brain [19], as the diagram overleaf shows.
However, homocysteine, unlike amyloid, is actually causal. That is, lowering homocysteine with B vitamins, stops the accelerated brain shrinkage, stops the cognitive decline and memory loss. That is consistent with a disease-modifying treatment and possibly the only thing, which from the current evidence, could be said to be causal. It is possible that some of its benefit is in lowering p-tau. There’s not enough research yet to say more than that at this point in time.
Of course, the ‘tauists’ know this and most of the drugs being developed try to do what lowering homocysteine does. Building on a few discoveries that ‘methylene blue’ [20], a methylated colouring, interfered with the formation of p-tau, and that the amino acid cysteine is involved in tau accumulation [21], drugs such as HMTM (Hydromethylthionine mesylate) exploit this bit of chemistry. Lack of B vitamins messes up methylation and homocysteine accumulates, leading to more p-tau formation. (Homocysteine is made from the amino acid methionine, which can also be turned into the amino acid cysteine, then glutathione – see Chapter 6). It is unlikely these drugs will have a substantial clinically significant effect, and much less so than lowering homocysteine. No doubt they will have adverse effects to factor in. But it won’t stop the drive to get such tau accumulation- inhibitor (TAI) drugs to market. Of course, it would be easier to just lower homocysteine with inexpensive and safe B vitamins, but these cannot be patented and hence cannot generate the profit pharma companies are looking for.
If treatment was really being driven by science, everyone would already be shouting about homocysteine lowering B vitamins (see Chapter 6). One senior representative of a pharma company told Professor David Smith, whose research on homocysteine is par excellence, that homocysteine lowering B vitamins would be a ‘multi-billion blockbuster drug if it could be patented’. But therein lies the problem
That doesn’t mean there won’t be other causes. Not everyone who develops Alzheimer’s has high homocysteine levels. There are other natural processes and compounds that can become damaging if they get out of balance. For instance, oxidants and inflammation protect against injury and infection but can damage mitochondria – the so called ‘energy’ factories inside every brain – if levels get too high. The effects of insulin resistance and damaged glucose control are similar.
Diabetes and dementia are strongly linked, the first doubling the risk of the second. [22] In truth, both homocysteine, which is a measure of a vital process called methylation, oxidation, insulin resistance and inflammation all affect the mitochondria. One clue for inflammation being involved relates to the finding that those with rheumatoid arthritis using heavy duty anti-inflammatory drugs have less risk for Alzheimer’s.[23]
These are some of the fruitful avenues that have been explored and show real promise. But they have all largely been ignored because of the unhealthy obsession by Pharma, the Alzheimer’s societies and government funding bodies on amyloid and tau. They will be explored in subsequent chapters.
In Food for the Brain’s model of dementia, glycation, oxidation, methylation and the vital role of brain fats, which actually build the brain, are central. I call them the ‘four horsemen of the mental health apocalypse’. The discovery that the homocysteine lowering B vitamins and omega-3 are co-dependent and together, dramatically slow brain shrinkage and improve cognitive function much better than any amyloid or p-tau treatment to date, is of major importance. Yet, this has been largely ignored by the blinkered Alzheimer’s establishment. So, next time you are asked to donate to Alzheimer’s charities ask them if any of the money is being spent on amyloid or p-tau. If it is, I’d suggest politely declining. If instead they are funding research into oxidation, inflammation, homocysteine, insulin or mitochondrial function, then that’s a much better sign that your money is being put to good use.
However, just focusing on one of these avenues may be misguided. It is based on the current paradigm of medical research – find the thing that is causing the disease, then ‘cure’ that. This assumes there is one cause and therefore one treatment. Of course, this is what you need for a drug to make money.
Let’s take homocysteine as an example. Not everyone who develops dementia or Alzheimer’s has high homocysteine. According to research at the US National Institutes of Health, it accounts for 22% of the risk.[24] Those who do have high homocysteine will reliably develop dementia and lowering it reliably reduces their cognitive decline. So, high homocysteine is a CAUSE, but not the only cause. Insulin resistance leads to diabetes and increases the risk for dementia. So, insulin resistance, driven by too much sugar and refined carbohydrates, is probably a cause, but not the only one. There isn’t enough evidence yet to declare ‘cause’, but the evidence that exists certainly points that way.
There is a different way of thinking and researching called ‘systems-based’ science. Much like the straw that breaks the camel’s back, this approach presumes there are a number of conditions, not just one, that can result in a disease such as Alzheimer’s or dementia. After all, a stroke or head injury can be a cause of cognitive decline, even if you don’t have high homocysteine or blood sugar problems. (It could be that a potential causal mechanism that ties these together is cerebrovascular dysfunction – disturbed blood supply to the brain. High homocysteine, by the way, increases risk of this by 17-fold [25]).
In my book Upgrade Your Brain, which gives all the referenced studies for statements made here, I argue that every known risk factor or biomarker for cognitive decline, dementia or Alzheimer’s affects either the structure, the function or the utilisation of the neuronal network and that it is combinations of these that crank up risk and ultimately brain pathology.
It’s like saying five critical things have to work for your car to move forward and not crash. Tyre pressure good, brakes working, enough gas, oil to lubricate the engine and water to cool it. If any one of these is completely broken, the car stops or crashes. If two are not working well, such as low oil and low water, the car grinds to a halt. If the brakes aren’t working, you crash.
We tend to think this way in nutrition and lifestyle medicine. It’s the combination of insults such as high sugar intake, too many fried foods, lack of vegetables, too much alcohol and smoking, that breaks the camel’s back. That heart attack is the ‘perfect storm’ of several underlying factors.
This systems-based approach isn’t popular in science and very few funders ever put up money to fund this kind of research. Usually, a funder wants to fund one stream of research, possibly a clinical trial of one approach, in the belief that this one factor is the key and a great discovery will be made. The reality is that it is usually combinations of factors that drive risk, with the manifestation of the disease itself being the ‘broken back’. Pollution, for example, is a risk factor for dementia … but not in those with good vitamin B6, folate or B12 status [26], which are the three B vitamins needed for methylation, indicated by lower homocysteine. Methylation is a major mechanism in the body, used to detoxify pollutants and toxins.
This is where Food for the Brain’s approach is unique. By collecting data from people like you who have both taken the Cognitive Function Test and completed the COGNITION questionnaire, and keep doing so, we can look at what drives cognitive function up and down. In other words, what ‘breaks the camel’s back’ or alternatively, makes it ‘strong’. This kind of complex systems-based science has become possible due to big data gathering (such as we are doing), advances in complex statistics, computer power and programming AI algorithms. Our Head of Science, Associate Professor Tommy Wood, is an expert in this kind of approach to neuroscience.
It is, I believe, the future and why we will probably find no single primary cause for Alzheimer’s, and certainly not amyloid or p-tau, but combinations of diet and lifestyle and other factors that create the tipping point that leads to dementia. Then, we will have the means to prevent this tipping point from ever being reached. In other words, we may discover that prevention is the cure.


Food for the Brain is a not-for-profit educational and research charity that offers a free Cognitive Function Test and assesses your Dementia Risk Index to be able to advise you on how to dementia-proof your diet and lifestyle.
By completing the Cognitive Function Test you are joining our grassroots research initiative to find out what really works for preventing cognitive decline. We share our ongoing research results with you to help you make brain-friendly choices.
Please support our research by becoming a Friend of Food for the Brain.
References:
3. Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015 Jun;18(6):794–799. doi: https://doi.org/10.1038/nn.4017 (Also see references and full discussion in Chapter 8 of How Not to Study a Disease, K. Herrup, MIT Press. Lopez OL, et al. Association Between β-Amyloid Accumulation and Incident Dementia in Individuals 80 Years or Older Without Dementia. Neurology. 2024 Jan 23;102(2):e207920.)
4. Salloway S, et al. Dominantly Inherited Alzheimer Network–Trials Unit. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021 Jul;27(7):1187–1196. doi: https://doi.org/10.1038/s41591-021-01369-8 Epub 2021 Jun 21.
5. Volloch V, et al. Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Med Sci (Basel). 2018 Jun 2;6(2):45. doi: https://doi.org/10.3390/medsci6020045
6. van Dyck CH, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023 Jan 5;388(1):9–21. doi: https://doi.org/10.1056/NEJMoa2212948 Epub 2022 Nov 29.
7. Walsh S, et al. Lecanemab for Alzheimer’s disease. BMJ. 2022;379:o3010. doi: https://doi.org/10.1136/bmj.o3010
15. Ackley SF, et al. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021 Feb 25;372:n156. doi: https://doi.org/10.1136/bmj.n156 Erratum in: BMJ. 2022 Aug 30;378:o2094.
10. Smith AD. Anti-amyloid trials raise scientific and ethical questions. BMJ. 2021;372:n805. doi: https://doi.org/10.1136/bmj.n805
11. Smith AD. Why are drug trials in Alzheimer’s disease failing? Lancet. 2010;376:1466. doi: https://doi.org/10.1016/S0140-6736(10)61994-0
12. Sims JR, et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023 Aug 8;330(6):512–527. doi: https://doi.org/10.1001/jama.2023.13239
14. Cummings J, et al. Alzheimer’s disease drug development pipeline: 2024. Alzheimers Dement (N Y). 2024 Apr 24;10(2):e12465. Dutch. doi: https://doi.org/10.1002/trc2.12465
19. Smith AD, Refsum H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu Rev Nutr. 2016 Jul 17;36:211–239. doi: https://doi.org/10.1146/annurev-nutr-071715-050947; see also Li JG, Chu J, Barrero C, Merali S, Pratico D. Homocysteine exacerbates β-amyloid, tau pathology, and cognitive deficit in a mouse model of Alzheimer’s disease with plaques and tangles. Ann Neurol. 2014;75:851–63; doi: https://doi.org/10.1002/ana.24166; see also Shirafuji N, et al. Homocysteine Increases Tau Phosphorylation, Truncation and Oligomerization. Int J Mol Sci.2018 Mar 17;19(3):891. doi: https://doi.org/10.3390/ijms19030891; see also Bossenmeyer-Pourié C, et al. N-homocysteinylation of tau and MAP1 is increased in autopsy specimens of Alzheimer’s disease and vascular dementia. J Pathol. 2019 Jul;248(3):291–303. doi: https://doi.org/10.1002/path.5254 Epub 2019 Mar 19.
20. Wischik CM, et al. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996 Oct 1;93(20):11213–8. doi: https://doi.org/10.1073/pnas.93.20.1121
21. Al-Hilaly YK, et al. Cysteine-Independent Inhibition of Alzheimer’s Disease-like Paired Helical Filament Assembly by Leuco-Methylthioninium (LMT). J Mol Biol. 2018 Oct 19;430(21):4119–4131. Epub 2018 Aug 16. Doi: https://doi.org/10.1016/j.jmb.2018.08.010
22. Arvanitakis Z, et al. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol.2004 May;61(5):661–666. doi: https://doi.org/10.1001/archneur.61.5.661; see also Yaffe K, et al. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology. 2004 Aug 24;63(4):658–663. doi: https://doi.org/10.1212/01.WNL.0000134665.93885.71; see also Tiehuis AM, et al. Diabetes Increases Atrophy and Vascular Lesions on Brain MRI in Patients With Symptomatic Arterial Disease. Stroke. 2008 May;39(5):1600–1603. doi: https://doi.org/10.1161/STROKEAHA.107.502963; see also Samaras K, et al. The impact of glucose disorders on cognition and brain volumes in the elderly: the Sydney Memory and Ageing Study. AGE. 2014;36(2):977–993. doi: https://doi.org/10.1007/s11357-013-9585-3; see also Mortby ME, et al. High ‘normal’ blood glucose is associated with decreased brain volume and cognitive performance in the 60s: the PATH through life study. PLoS One. 2013 Sep 4;8(9):e73697.
doi: https://doi.org/10.1371/journal.pone.0073697; see also Crane PK, et al. Glucose levels and risk of dementia. N Engl J Med. 2013 Aug 8;369(6):540–548. doi: https://doi.org/10.1056/NEJMoa1215740; see also Luchsinger JA, et al. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004 Oct 12;63(7):1187–1192. doi: https://doi.org/10.1212/01.wnl.0000140292.04932.87; see also Abbatecola AM, et al. Insulin resistance and executive dysfunction in older persons. J Am Geriatr Soc. 2004 Oct;52(10):1713–1718. doi: https://doi.org/10.1111/j.1532-5415.2004.52466.x; see also Ye X, et al. Habitual sugar intake and cognitive function among middle-aged and older Puerto Ricans without diabetes. Br J Nutr. 2011 Nov;106(9):1423–1432. doi: https://doi.org/10.1017/S0007114511001760; see also Power SE, et al. Dietary glycaemic load associated with cognitive performance in elderly subjects. Eur J Nutr.2015 Jun;54(4):557–568. doi: https://doi.org/10.1007/s00394-014-0737-5; see also Seetharaman S, et al. Blood glucose, diet-based glycemic load and cognitive aging among dementia-free older adults. J Gerontol A Biol Sci Med Sci. 2015 Apr;70(4):471–479. doi: https://doi.org/10.1093/gerona/glu135; see also Taylor MK, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017 Dec;106(6):1463–1470. doi: https://doi.org/10.3945/ajcn.117.162263; see also Gentreau M, et al. High Glycemic Load Is Associated with Cognitive Decline in Apolipoprotein E ε4 Allele Carriers. Nutrients. 2020 Nov 25;12(12):3619. doi: https://doi.org/10.3390/nu12123619
23. Xie W, et al. Association between disease-modifying antirheumatic drugs for rheumatoid arthritis and risk of incident dementia: a systematic review and meta-analysis. RMD Open. 2024 Feb 27;10(1):e004016. doi: https://doi.org/10.1136/rmdopen-2023-004016
24. Beydoun MA, et al. Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health. 2014 Jun 24;14:643. doi: https://doi.org/10.1186/1471-2458-14-643
25. Teng Z, et al. Cerebral small vessel disease mediates the association between homocysteine and cognitive function. Front Aging Neurosci. 2022;14:868777. doi: https://doi.org/10.3389/fnagi.2022.868777
26. Chen C, et al. B vitamin intakes modify the association between particulate air pollutants and incidence of all-cause dementia: Findings from the Women’s Health Initiative Memory Study. Alzheimers Dement. 2022 Nov;18(11):2188–2198. doi: https://doi.org/10.1002/alz.12515 Epub 2022 Feb 1.
